O código CID E76.0 refere-se à mucopolissacaridose do tipo I, uma doença genética hereditária pertencente ao grupo das doenças de armazenamento lisossomal. Essa condição resulta na deficiência da enzima alfa-L-iduronidase, que leva ao acúmulo de mucopolissacarídeos (glicosaminoglicanos) em tecidos, órgãos e sistemas do corpo. A mucopolissacaridose tipo I apresenta um espectro de severidade, incluindo a forma grave conhecida como Síndrome de Hurler, e formas mais leves como Scheie e intermédia, muitas vezes relacionada à deficiência parcial da enzima. Os sintomas geralmente incluem disfunções respiratórias, deformidades ósseas, hipertrofia de órgãos, impacto cognitivo variável, dificuldades de mobilidade, problemas de visão e audição, além de alterações na pele e dentes. O diagnóstico é feito por testes laboratoriais que medem níveis de enzimas e por exames genéticos. O tratamento envolve reposição enzimática, cirurgias ortopédicas e suporte multidisciplinar. A prevenção se baseia no aconselhamento genético e em diagnósticos pré-natais.
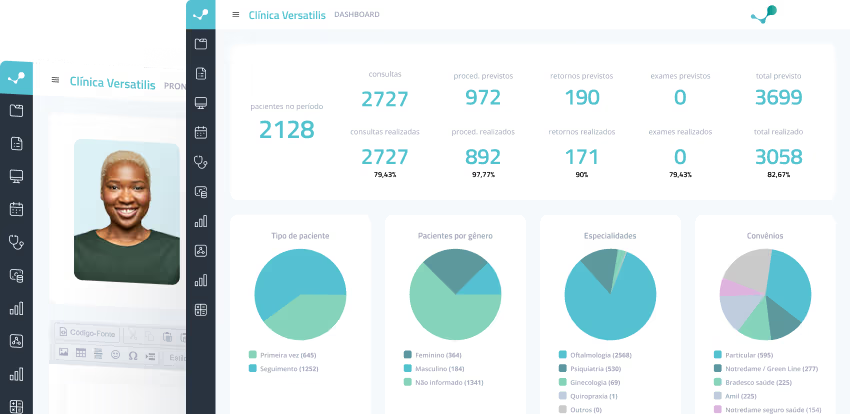

Versatilis System - O software definitivo para gestão de clínicas e consultórios.
